



A Discourse on Residue Limits
Learn to calculate residue limits in cleaning validation, ensuring safety, preventing contamination, and meeting compliance in pharma facilities.
This detailed guide provides an in-depth look into the calculation of residue limits, a critical component for preventing cross-contamination in shared facilities.
Deep Dive into Cleaning Limits
Dosage Criteria: An In-depth Exploration
In pharmaceutical manufacturing, the Dosage Criteria method establishes acceptable residue levels based on the Minimum Daily Dose (MDD) of an active ingredient (API) and a predefined Safety Factor (SF). The Safety Factor typically set at 0.001 for solids, is derived from toxicological and exposure assessments, intended to ensure an adequate margin of safety.
Calculation:

In this formula:
MDD of API is the lowest clinically effective dose of the active ingredient per day.
SF (Safety Factor) introduces a buffer to account for uncertainties in toxicological data and inter-individual differences.
LDD (Largest Daily Dose) refers to the highest anticipated daily intake of the subsequent product manufactured using the same equipment.
The key mistake to avoid here is confusing the MDD of the API with the LDD of the product, which includes excipients alongside the API.
ADE/PDE Criteria: Comprehensive Breakdown
The ADE (Acceptable Daily Exposure) and PDE (Permitted Daily Exposure) criteria provide an alternative approach focusing on the maximum safe exposure level to the residue over a lifetime without adverse effects.
Calculation:
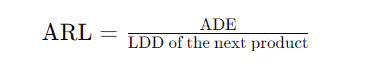
Here:
ADE (mg/person/day) is calculated based on toxicological evaluation, representing a safe exposure level over a lifetime.
LDD includes the entire formulation, not just the active component.
Setting Default ARL Limits: Beyond the Basics
In pharmaceutical cleaning validation, establishing Default Acceptable Residue Limits (ARLs) is crucial for maintaining consistent safety standards across different products and manufacturing scenarios. The concept of a default ARL provides a 'ceiling' limit that ensures even if calculated limits based on Dosage or ADE/PDE criteria exceed this threshold, the default limit prevails to maintain strict safety margins.
Typically, pharmaceutical companies adopt a standard default ARL, often set at 10 parts per million (ppm). This default limit is derived from industry best practices and regulatory guidelines, serving as a universal benchmark to prevent excessively high residue levels regardless of the specific characteristics of the drug product or the active pharmaceutical ingredient (API) involved.
Importance of Default Limits
The establishment of a default limit is particularly significant in scenarios where theoretical calculations yield unreasonably high permissible residue amounts, which could potentially compromise patient safety or product quality. By setting a default limit, companies ensure a consistent safety buffer is maintained, reflecting a conservative approach to patient safety and regulatory compliance.
Application in Cleaning Validation
During the cleaning validation process, if the calculated ARL for a particular API based on Dosage or ADE/PDE criteria exceeds the default limit (for example, if the Dosage-based ARL calculation results in 15 ppm), the default ARL of 10 ppm is applied instead. This approach ensures that the final residue limits enforced are within a safe and acceptable range, regardless of the variations in API potency, dosage form, or production processes.
The default ARL is particularly vital in shared facilities where multiple products are manufactured, as it establishes a uniform standard that applies across different product lines and equipment. This uniformity is essential for simplifying cleaning validation protocols and ensuring comprehensive contamination control in multi-product environments.
By adhering to these default limits, pharmaceutical manufacturers underscore their commitment to safety and compliance, while also addressing the inherent variability and complexities associated with cleaning validation and residue analysis.

Mastery of MAC and Surface Area Limits
MAC Calculation:
For shared pharmaceutical equipment, calculating the Maximum Allowed Carryover (MAC) involves determining the highest acceptable amount of residue from the previous product:

This ensures that the subsequent batches will not be contaminated beyond safe levels.
Surface Area Limit (SAL) Calculation:

The SAL provides a concrete figure for the allowable residue per unit area, facilitating targeted cleaning and validation efforts.
Advanced Analysis of Swab and Rinse Limits
Swab and rinse testing are critical components of the cleaning validation process, determining the physical removal of contaminants from equipment surfaces.
Swab Limits are calculated based on the smallest SAL applicable to the shared equipment, adjusted by the swabbed area and solvent volume:

This considers the physical removal efficiency of the swabbing process and the solvent's capacity to capture and hold residues.
Rinse Limits are similarly derived but focus on the volume of solvent used to rinse the equipment surface, ensuring all residues are effectively removed from larger areas or areas not accessible by swabbing:

The selection of solvents and methodologies for determining swab and rinse areas are tailored to the chemical nature of the residues and the material characteristics of the equipment, ensuring effective residue removal while avoiding equipment damage.

Evaluating Worst Case Products with Precision
When identifying worst-case products for equipment cleaning validation, we consider multiple factors to ensure the cleaning process is robust and comprehensive. The objective is to ensure the chosen product for validation is representative of the most challenging cleaning scenario.
Determining Factors:
Solubility: Products are categorized based on their solubility in various solvents. For example, between a product that is 'soluble', one that is 'sparingly soluble', and another that is 'practically insoluble', the 'practically insoluble' product would be considered the worst case as it is the hardest to clean.
Chemical properties: Consideration of the API's chemical properties, including its adherence to surfaces, degradation products, and interaction with cleaning agents, informs the worst-case scenario.
Dosage form and potency: Highly potent compounds or those in a sticky or viscous form pose greater cleaning challenges.
Equipment contact surface area: A product processed in equipment with a large contact surface area or complex design may be considered worst-case due to the difficulty in cleaning all surfaces effectively.
Evaluation Process:
List all products sharing the equipment and their relevant characteristics (solubility, potency, form).
Rank each product based on the difficulty of removing their residues—considering solubility, potency, and interaction with cleaning agents.
Determine which product has the highest risk associated with cross-contamination due to residue carryover. This involves a combination of factors including but not limited to toxicity (ADE/PDE values) and residue tenacity.
Validate cleaning processes using the identified worst-case product, ensuring that cleaning procedures are effective against the most challenging conditions.
This comprehensive approach ensures that cleaning validation protocols are stringent enough to manage the highest risk scenarios, thereby ensuring safety across all manufactured products.
Common Mistakes and Misconceptions
In calculating residue limits and conducting cleaning validation, common mistakes can compromise the integrity of the process:
Misinterpreting LDD (Largest Daily Dose): Confusing this with the maximum daily dose of the API instead of considering the total product formulation.
Inadequate safety margins: Failing to apply an appropriate safety factor (SF) for different product forms or overlooking patient population differences.
Incorrect solubility assumptions: Misclassifying the solubility of APIs leading to underestimating the difficulty of cleaning.
Neglecting equipment complexity: Not considering all surfaces, nooks, and crannies that may harbor residues, especially in multifunctional or intricate equipment.
Overlooking cross-contamination routes: Ignoring indirect contamination paths, such as airborne particles or fluid carryover.
Addressing these mistakes requires a thorough understanding of the principles underpinning cleaning validation and an attention to detail in the application of those principles.

Regulatory Perspectives and Compliance
The regulatory landscape for cleaning validation is governed by guidelines from bodies such as the FDA, EMA, and WHO. These guidelines emphasize a science-based, risk-managed approach, stressing the importance of:
Validation Master Plan: A comprehensive document outlining the approach to cleaning validation, including protocols, methodologies, and acceptance criteria.
Risk Assessment: Evaluating the risk of cross-contamination based on product toxicity, equipment complexity, and manufacturing processes.
Documentation and Traceability: Maintaining detailed records of cleaning validation studies, procedures, and outcomes to ensure traceability and compliance.
Continuous Monitoring and Improvement: Regularly reviewing and updating cleaning protocols based on new products, equipment, or regulatory standards.
By aligning with these regulatory perspectives, manufacturers ensure not only compliance but also the safety and efficacy of their products.









.avif)






