Cleaning Validation Guidelines 2025

In today’s fast-evolving pharmaceutical industry, cleaning validation guidelines are no longer just regulatory checkboxes—they are strategic tools for ensuring patient safety, product quality, and regulatory compliance across the entire manufacturing lifecycle.
As global agencies like the FDA, EMA, MHRA, and WHO update expectations for equipment cleaning procedures, sampling methods, and risk-based validation, pharmaceutical manufacturers must evolve their cleaning validation programs to stay compliant and competitive.


This 2025 guide synthesizes the latest global regulations, best practices, and digital innovations into a unified strategy for building a science-driven, audit-ready cleaning validation process—from manual cleaning procedures to continuous monitoring.
Whether you're validating fluid bed dryer bags, managing residue removal from shared manufacturing equipment, or digitizing your validation protocols, this guide gives you the tools to lead with confidence and stay inspection-ready—every time.
Why Cleaning Validation Guidelines for the Equipment Cleaning Process Matter More Than Ever?
A New Era of Regulatory Accountability in the Cleaning Process
Global regulators now require scientifically justified cleaning validation procedures that do more than remove visible residues—they must ensure consistent, measurable elimination of chemical, microbial, and cross-contaminant risks.
Agencies like the FDA emphasize written procedures, validated analytical methods (such as TOC and swab sampling), and equipment surface verification. Meanwhile, the EMA mandates Health-Based Exposure Limits (HBELs) and statistically derived residue limits based on the maximum daily dose of active pharmaceutical ingredients (APIs).
Strategic Implications for Pharma Facilities
An effective cleaning validation lifecycle contributes directly to:
- Patient safety through reduction of cross contamination risk
- Operational efficiency by avoiding reworks and batch rejections
- Inspection consistency and global compliance across facilities
In complex, multi-product environments with long transfer lines, rotating spray devices, or shared processing equipment, these guidelines are crucial to validate cleaning processes under worst-case conditions.
Pro Tip: Don’t overlook non-product contact surfaces (like HVAC vents, wash tanks, and walls). Regulators increasingly expect complete cleaning verification across all zones—visible and hidden.
FDA Cleaning Process Validation Guidelines 2025 for Effective Cross-Contamination Prevention
The U.S. Food and Drug Administration (FDA) continues to lead the charge in evolving cleaning validation guidelines—pushing manufacturers toward risk-based, data-driven, and digitally supported cleaning programs.
What the FDA Expects in 2025
Under 21 CFR Part 211.67, FDA regulations now place greater emphasis on:
- Scientifically Justified Validation Protocols
Cleaning validation protocols must include clearly defined sampling procedures, analytical methods, acceptance criteria, and rationale for residue limits based on the product’s normal therapeutic dose. - Risk-Based Cleaning Validation Programs
Validation must address worst-case contamination scenarios, especially in facilities with shared equipment or manual cleaning procedures. Risk assessments should consider product potency, solubility, and equipment complexity. - Advanced Analytical Methods
The FDA encourages using a combination of swab testing, TOC analysis, and rinse samples to verify residue removal. These methods should be validated for- Detection limit
- Specificity
- Recovery efficiency
- Continuous Process Verification
Post-validation, cleaning must be monitored through routine cleaning records, trend data, and digital logs to ensure ongoing control and consistency.
Key Red Flags (and How to Avoid Them)
FDA inspections frequently cite deficiencies like:
- Incomplete or outdated validation protocols
- Lack of defined sampling locations
- Absence of a final validation report
- Overreliance on visual inspection as sole acceptance criteria


To mitigate these risks, use software that automates protocol management, flags missing data, and standardizes documentation across your manufacturing system.
How to Stay FDA Audit-Ready
To ensure compliance under 2025 guidelines:
- Standardize written cleaning procedures with clear rationale
- Validate analytical methods for both direct surface sampling and rinse testing
- Train personnel on manual cleaning techniques and sampling methods
- Use digital tools to enable:
- Real-time tracking of cleaning activities
- Automated alerts for revalidation triggers
- Centralized audit logs for inspectors and other FDA personnel
Pro Tip: Want to streamline documentation, CAPA workflows, and compliance tracking?
Tools like Leucine’s Cleaning Validation Software automate critical steps, reduce risk, and improve inspection outcomes
EMA Cleaning Validation Guidelines & HBELs
The European Medicines Agency (EMA) brings a more toxicology-driven, exposure-based approach to cleaning validation—demanding precision in setting residue limits, especially in multi-product facilities.
Annex 15 of the EU GMP Guide outlines a cleaning validation framework that focuses on patient safety through scientifically calculated Health-Based Exposure Limits (HBELs).
EMA’s Core Requirements for 2025
To comply with EMA standards, your cleaning validation program should include:
- HBEL-Based Residue Limits
Residue limits must be derived using toxicological data such as:- Permitted Daily Exposure (PDE)
- Maximum Allowable Carryover (MAC)
- Worst-case product scenarios in shared equipment
- Statistical and Historical Data Integration
EMA encourages blending historical dosage data with modern statistical tools to justify cleaning thresholds. This means moving beyond visual inspection and arbitrary limits. - Lifecycle Management and Revalidation
Periodic reassessment of residue limits is required, especially after:- Changes in formulation
- New equipment introduction
- Revised HBELs based on updated toxicology data
- Robust Documentation
All cleaning validation activities—protocols, sampling methods, HBEL justifications, and test data—must be documented with traceable digital records.
What Sets EMA Apart?
Unlike the FDA, which accepts visual inspection as part of its acceptance criteria, EMA insists that quantified residue limits be calculated from toxicological principles—visual checks alone are not acceptable.
EMA’s cleaning validation lifecycle approach also leans heavily into:
- Continuous improvement
- Periodic protocol reassessment
- Regulatory inspection preparedness through detailed audit trails
How to Stay EMA Audit-Ready
To align with EMA’s updated guidance:
- Perform toxicological evaluations to establish residue limits
- Apply HBELs to all relevant product-contact and non-product-contact surfaces
- Use statistical process controls to monitor cleaning effectiveness
- Adopt digital tools that support:
- HBEL/MAC tracking
- Document version control
- Trigger-based revalidation workflows
Pro Tip: Leucine’s platform helps automate HBEL-based calculations, track exposure data, and unify your documentation into a single audit-ready system.
MHRA Guidelines for Risk-Based Cleaning Validation
The UK’s Medicines and Healthcare Products Regulatory Agency (MHRA) closely aligns with EMA’s science-based approach—but with an added focus on real-time digital documentation and proactive risk management.
In a post-Brexit regulatory landscape, MHRA has intensified its expectations for digital compliance infrastructure and continuous verification.
MHRA’s Key Cleaning Validation Expectations
Here’s what the MHRA emphasizes for 2025:
- Enhanced Risk Assessments
Validation must evaluate factors like:- Equipment design and cleanability
- Compatibility of cleaning agents
- Frequency of routine cleaning cycles
- Potential for cross contamination
- Digital Record-Keeping Systems
MHRA prefers electronic logs, real-time status dashboards, and secure, audit-ready data trails. Handwritten logs or disconnected systems raise inspection flags. - Training and Process Ownership
Personnel responsible for manual cleaning, sampling procedures, and visual inspections must receive role-specific training. Audit failures often stem from inconsistent execution, not bad SOPs.
How to Stay MHRA Audit-Ready
To align with MHRA standards:
- Use digital systems for scheduling revalidations, deviation tracking, and audit reporting
- Conduct frequent internal audits and CAPA reviews
- Ensure every cleaning validation protocol includes traceable records of:
- Sampling locations
- Acceptance criteria
- Analytical test data
- Integrate real-time monitoring tools for equipment cleaning processes, especially in high-risk manufacturing zones
ANVISA – Cleaning Validation and Documentation in Brazil Validation and Documentation in Brazil Overall Expectation:
ANVISA, Brazil’s National Health Surveillance Agency, mirrors WHO-aligned cleaning validation expectations but layers on additional controls tailored to high-volume, multi-product environments common in the region.
With a strong emphasis on documentation traceability, risk-based cleaning procedures, and local GMP standards, ANVISA’s guidelines are critical for any pharma facility operating in or exporting to Brazil.
ANVISA’s Key Cleaning Validation Guidelines
To meet ANVISA’s 2025 expectations, your program should include:
- Localized Risk Assessments
Validation protocols must account for:- Diverse formulations and shared equipment
- Unique cleaning agents used in Brazil
- Facility-specific challenges like long equipment runs or manual cleaning procedures
- Exhaustive Documentation & Traceability
Every cleaning activity must be traceable:- From sampling method → to test data → to final validation report
- Include direct sampling, rinse samples, equipment surface logs, and cleaning agent residues
- Periodic Review and Revalidation
Validation is not “one and done.” ANVISA mandates:- Trigger-based revalidations (e.g., equipment changes, formulation updates)
- Regular internal reviews and performance metrics to ensure long-term cleaning efficacy
How to Stay ANVISA Audit-Ready
To ensure inspection consistency under ANVISA:
- Define sampling plans for manual and automated cleaning processes
- Include both product-contact and non-contact surfaces in validation scope
- Track cleaning agent residue removal with validated MAC calculations
- Use software that enables:
- Trigger-based revalidation workflows
- Audit logs customized for ANVISA format
- Support for Brazilian GMP formatting and reporting standards
Pro Tip: Leucine enables site-specific risk mapping and localized compliance tracking to meet ANVISA’s detailed documentation standards without adding manual workload.
PIC/S – Guidelines for Shared Facilities & Worst-Case Scenarios
The Pharmaceutical Inspection Co-operation Scheme (PIC/S) brings together over 50 international regulatory agencies, creating one of the most harmonized sets of cleaning validation guidelines in the pharmaceutical world. Its emphasis? Risk-based strategies, worst-case evaluations, and global consistency.
For manufacturers operating across borders, compliance with PIC/S means smoother audits, fewer remediation cycles, and better alignment with EMA, TGA, Health Canada, and others.
Core PIC/S Requirements for Cleaning Validation
PIC/S guidelines (notably PI-046-1 and Aide-Memoire PI-052-1) require:
- Worst-Case Scenario Validation
You must prove that your cleaning methods can remove residues under the most challenging product conditions—low solubility, high toxicity, or sticky formulations. - Validated Sampling & Analytical Methods
Visual inspection alone is not sufficient. Use a combination of:- Swab sampling
- Rinse samples
- Total Organic Carbon (TOC) analysis
- HPLC or other validated methods
These must be supported by scientifically justified acceptance criteria and recovery studies.
- Lifecycle-Based Cleaning Validation
PIC/S mandates a full lifecycle approach, from initial validation to periodic requalification. This includes:- Real-time documentation
- Continuous verification
- Integration with your quality assurance system
Prohibition of Trial-and-Error
Repeated cleaning until "clean enough" is explicitly prohibited. Your cleaning validation process must work first time, every time, under validated conditions.
How to Stay PIC/S-Compliant in Multi-Product Environments
In facilities with long transfer lines, shared processing equipment, or manual cleaning routines, PIC/S requires:
- Justification for equipment grouping during validation
- Rigorous documentation of residue removal performance under worst-case conditions
- Automated systems for revalidation scheduling and trend tracking
Pro Tip: Leucine supports PIC/S-aligned lifecycle validation with:
-Real-time process verification
-Sampling workflows and test data traceability
-Interactive dashboards that support cross-site validation consistency
ASTM Standards – Science-Based Validation Built on ICH Principles
The American Society for Testing and Materials (ASTM) offers one of the most scientifically grounded frameworks for cleaning validation through its ASTM E3106-18e1 standard. This guideline aligns closely with ICH Q9 (Quality Risk Management) and Q10 (Pharmaceutical Quality System)—focusing on statistical rigor, risk assessment, and global harmonization.
For pharma manufacturers aiming to future-proof their validation program, ASTM sets the gold standard for integrating quantitative science with real-world cleaning practices.
Core ASTM E3106 Expectations
- Data-Driven Cleaning Validation Protocols
Cleaning validation procedures should be designed around:- Risk-based product and equipment selection
- Statistically justified acceptance criteria
- Measurable analytical methods with proven detection limits
- Statistical Evaluation of Cleaning Effectiveness
Unlike basic pass/fail validation, ASTM promotes:- Trend analysis over time
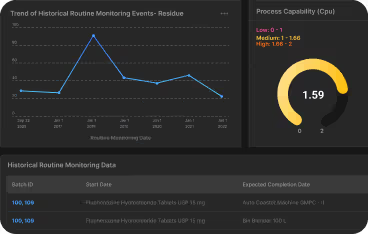
- Process capability indices (Cp/Cpk)
- Monte Carlo simulations or similar risk modeling techniques
- Alignment with Global Standards
The ASTM approach is structured to harmonize with FDA, EMA, and WHO requirements—providing a universal framework for multi-site or multi-national validation efforts.
How to Implement ASTM-Based Cleaning Validation
To meet ASTM expectations in 2025:
- Use quantitative data to justify limits (avoid visual-only assessments)
- Track validation outcomes using control charts or trending dashboards
- Standard sampling procedures for cleaning agents, equipment surfaces, and manual cleaning processes
- Ensure your system can manage:
- Test data analytics
- Risk scoring across multiple products and lines
- Version-controlled reports
Pro Tip: Leucine’s platform supports ASTM-aligned cleaning validation by embedding:
-Statistical dashboards for performance trending
-Automated test result logging and threshold flags
-Lifecycle tracking with deviation linkage
PDA and ISPE: Cleaning Validation Lifecycle, Equipment Cleaning SOPs & Cross-Contamination Control
Two of the most influential industry organizations—the Parenteral Drug Association (PDA) and the International Society for Pharmaceutical Engineering (ISPE)—have shifted the cleaning validation conversation from a static event to a continuous lifecycle process.
Their guidelines emphasize a proactive, end-to-end strategy for cleaning validation that spans:
- Initial qualification
- Continuous monitoring
- Data-driven revalidation
- Audit-readiness throughout the product lifecycle
PDA & ISPE Best Practices for 2025
- Lifecycle Management
Cleaning validation must be treated like any other critical process—subject to periodic review, trending, and change control:- Ongoing monitoring (e.g., swab data over time)
- Cleaning SOPs updated with new materials or formulations
- Digital tracking of performance indicators
- Standardized Documentation & Reporting
Use structured templates for:- Cleaning validation protocols
- Deviation logs
- Final validation reports
These should be version-controlled, timestamped, and auditable across your manufacturing system.
- Cross-Functional Alignment
Collaboration is key. Validation should connect:- QA and QC teams
- Manufacturing and engineering
- Training and regulatory affairs
The result: fewer silos, faster investigations, and better compliance outcomes.
How to Operationalize PDA & ISPE Guidance
To apply their lifecycle-centric approach:
- Build a digital framework for routine cleaning and trigger-based revalidation
- Integrate environmental monitoring data with cleaning validation metrics
- Standardize document control to improve inspection consistency
- Schedule ongoing internal audits and training refreshers for all cleaning personnel
Pro Tip: These guidelines are ideal for scaling cleaning validation programs across multiple sites or CMOs—especially when digital coordination is key.
WHO – Worst-Case Cleaning, HBELs & Stability-Based Residue Limits
The World Health Organization (WHO) takes a no-compromise stance on cross-contamination control—especially for multi-product facilities and resource-constrained settings.
Its Good Manufacturing Practices (GMP) for Active Pharmaceutical Ingredients outline a globally respected framework built around worst-case evaluation, health-based residue limits, and ongoing verification of cleaning effectiveness.
Key WHO Cleaning Validation Principles
- Worst-Case Scenario Approach
Cleaning validation protocols must demonstrate effective residue removal in the most difficult, high-risk situations, including:- Highly potent APIs
- Low-solubility compounds
- Poorly accessible equipment surfaces (e.g., fluid bed dryer bags)
- Health-Based Exposure Limits (HBELs)
WHO requires residue limits be set using toxicological data like:- Permitted Daily Exposure (PDE)
- Maximum Safe Carryover (MSC)
- Maximum Safe Surface Residue (MSSR)
- Stability Data Integration
Manufacturers are expected to use stability studies to inform residue limits—adjusting them as new data on degradation or carryover emerges. - Combination of Sampling Methods
WHO supports multi-method validation strategies using:- Swab sampling
- Rinse testing
- Visual inspection (as supplementary only)
- Validated analytical methods such as TOC or HPLC
Continuous Verification & Lifecycle Management
Cleaning validation is not static—it must be maintained via routine monitoring, trend reviews, and risk-based revalidations.
How to Align with WHO Expectations
To maintain compliance with WHO guidelines:
- Use scientific evidence to justify cleaning procedures
- Integrate stability data when updating residue thresholds
- Validate cleaning of non-product-contact surfaces as part of the holistic process
- Establish a Cleaning Validation Master Plan (CVMP) that includes:
- Defined sampling locations
- Analytical method rationale
- Deviation protocols and CAPA triggers
Pro Tip: Leucine's lifecycle validation platform helps unify your master plan, track HBEL implementations, and maintain a defensible audit trail from cleaning event to inspection day.
Health Canada & TGA – Dynamic Revalidation and Digital Documentation
While not as frequently spotlighted as FDA or EMA, Health Canada and Australia’s TGA (Therapeutic Goods Administration) are increasingly important for manufacturers operating across North America, APAC, and global markets.
Both agencies are PIC/S members and follow WHO-aligned GMP principles—but add emphasis on localized compliance, agile revalidation, and digital readiness for evolving operations.
Key Requirements from Health Canada & TGA
- Dynamic Revalidation Mechanisms
Cleaning validation protocols must be updated dynamically—not just periodically. Triggers include:- New equipment or cleaning agents
- Process changes
- Batch or formulation deviations
- Revised maximum daily dose for APIs
- Real-Time Digital Documentation
Inspectors expect:- Timestamped logs of cleaning activities
- Final validation reports stored and accessible electronically
- Change control and deviation records tied to the cleaning validation lifecycle
- Risk-Based Validation Approach
Like EMA and MHRA, these agencies require robust risk assessments based on:- Equipment design
- Exposure risk
- Cross-contamination potential
- Ongoing Training & Internal Auditing
Training for trained personnel must be documented and current. Internal audits are required to validate cleaning processes and assess inspection consistency.
How to Maintain Audit-Readiness with Health Canada & TGA
To stay compliant:
- Implement trigger-based revalidation calendars
- Use a centralized digital system for logging validation results, updates, and approvals
- Document detailed sampling procedures, cleaning methods, and equipment cleaning steps
- Link risk assessments, acceptance criteria, and sampling locations across your entire cleaning validation lifecycle


Pro Tip: With Leucine, revalidations are no longer a scramble. The system auto-triggers reviews, connects cleaning activities to test data, and offers real-time visibility into compliance status.
Conclusion: Global Cleaning Validation Guidelines – Your Path to Strategic Compliance
In 2025, cleaning validation guidelines are more than compliance checklists — they’re your blueprint for regulatory excellence, product safety, and competitive advantage.
Across FDA, EMA, MHRA, WHO, PIC/S, ANVISA, TGA, and Health Canada, the message is consistent:
You need a risk-based, lifecycle-driven, digitally traceable cleaning validation program that can adapt to change, prove its effectiveness, and stand up to global audits.
Frequently Asked Questions