FDA Cleaning Validation: How AI Automation Prevents Industry-Wide 483 Compliance Failures

The Critical FDA Cleaning Validation Challenge
Pharmaceutical manufacturing faces unprecedented regulatory scrutiny around FDA cleaning validation processes. Recent industry data reveals that cleaning validation-related FDA 483 observations have surged by 45% in the past 18 months, with equipment cleaning violations under 21 CFR 211.67 ranking as the third most frequent observation across pharmaceutical facilities worldwide.
The regulatory landscape demands more than basic equipment cleaning procedures. Modern pharmaceutical companies need FDA cleaning validation systems that not only meet current 21 CFR Part 211.67 requirements but also anticipate evolving regulatory expectations for risk-based validation, health-based exposure limits (HBELs), and comprehensive cross-contamination prevention.
Recent FDA Cleaning Validation Compliance Trends:
- 52% escalation rate from 483 observations to Warning Letters when cleaning validation inadequacies persist
- Average 8-14 month delays in product approvals due to FDA cleaning validation compliance gaps
- $3-8M per batch potential financial impact from cross-contamination incidents
- Risk-based validation approaches becoming regulatory expectation, not just recommendation
This industry challenge presents both regulatory risk and competitive opportunity. Companies investing in modern, AI-powered FDA cleaning validation systems are achieving compliance excellence that prevents regulatory observations while accelerating time-to-market and reducing operational costs.
Understanding FDA Cleaning Validation Failure Patterns from Recent 483 Observations
Torrent Pharmaceuticals Limited: A Critical FDA Cleaning Validation Failure
FDA 483 Observation Details:
- Company: Torrent Pharmaceuticals Limited, Kadi, India (FEI: 3005029956)
- Issue Date: June 12, 2024
- Inspection Dates: June 3-12, 2024
- FDA Investigators: Pratik S. Upadhyay, DDC and Steven A. Brettler
- Key Observation: Inadequate cleaning procedures for non-dedicated manufacturing equipment failing to ensure removal of residual materials from previously manufactured drug products, risking cross-contamination
The Compliance Failure: Inadequate Equipment Cleaning Procedures
The regulatory observation at Torrent Pharmaceuticals highlighted a fundamental FDA cleaning validation failure that echoes across the pharmaceutical industry. Investigators found that the company's cleaning procedure for non-dedicated manufacturing equipment was inadequate to ensure complete removal of residual materials from previously manufactured drug products, creating significant cross-contamination risks.
This observation directly violates 21 CFR Part 211.67 (Equipment cleaning and maintenance) and represents one of the most critical compliance gaps in pharmaceutical manufacturing. Industry analysis shows that similar FDA cleaning validation failures have been identified across 89 pharmaceutical sites in 13 countries, indicating this is a systemic industry challenge.
Regulatory Context: Why FDA Cleaning Validation Compliance is Critical
21 CFR Part 211.67: The Equipment Cleaning Imperative
The regulatory requirements for 21 CFR Part 211.67 mandate that equipment used in manufacturing must be:
- Of appropriate design and adequate size for intended use
- Properly cleaned and maintained to prevent contamination
- Suitable for its intended use including effective cleaning procedures
- Protected from contamination when not in use
- Inspected for cleanliness before use
ALCOA+ Principles and FDA Cleaning Validation Integrity
Modern pharmaceutical FDA cleaning validation must align with ALCOA+ data integrity principles:
- Attributable: Clear ownership of cleaning validation activities and results
- Legible: Documentation supporting regulatory review and inspection readiness
- Contemporaneous: Real-time capture of cleaning validation data and deviations
- Original: Authentic residue testing data without manipulation
- Accurate: Precise limit calculations and scientifically justified acceptance criteria
- Complete: Comprehensive validation covering all equipment, products, and worst-case scenarios
- Consistent: Standardized methodology across all cleaning validation protocols
- Enduring: Permanent records supporting product lifecycle and regulatory decisions
- Available: Accessible validation data for inspection and business continuity
Root Cause Analysis: Why Traditional FDA Cleaning Validation Systems Fail
The Paper-Based Validation Trap
Most pharmaceutical companies still rely on legacy FDA cleaning validation approaches that cannot meet modern regulatory expectations:
Manual Validation Workflows:
- Validation teams spend 75% of their time on data collection rather than scientific analysis
- Critical connections between cleaning effectiveness and cross-contamination risks are missed
- Residue limit calculations lack systematic methodology and statistical rigor
- CAPA effectiveness from cleaning failures is inconsistently monitored
Disconnected Systems and Data Silos:
- Manufacturing cleaning data exists separately from analytical testing results
- Equipment history and product changeover data integration challenges
- Validation findings are not accessible for trending and risk assessment
- Knowledge management gaps lead to repeated validation protocol mistakes
Human Error and Resource Constraints:
- 65% of cleaning validation delays are due to resource availability rather than technical complexity
- Inconsistent validation quality depends on individual validator experience
- Documentation gaps create regulatory vulnerability during inspections
- Training inefficiencies result in procedural deviations and compliance failures
Business and Regulatory Consequences of FDA Cleaning Validation Failures
The cost of inadequate FDA cleaning validation extends far beyond regulatory citations:
Financial Impact:
- Batch holds and delays: Torrent-type failures can hold $3-8M worth of product per batch
- Cross-contamination incidents: Product recalls averaging $15-25M in direct costs
- Warning Letter progression: 52% of cleaning validation 483s escalate to enforcement actions
- Market access delays: Validation inadequacies delay approvals by 8-14 months
Operational Consequences:
- Supply chain disruption: Cleaning validation delays create customer delivery failures
- Resource drain: Manual validation processes consume 60-80 hours per protocol
- Compliance risk: Inadequate validation leads to repeat observations and enforcement
- Competitive disadvantage: Slower product launches compared to digitally advanced competitors
Calculate Your ROI from automated FDA cleaning validation processes.
Leucine CLEEN: The Modern Solution to FDA Cleaning Validation Compliance
AI-Powered FDA Cleaning Validation That Prevents 483 Observations
Leucine CLEEN is specifically designed to address the FDA cleaning validation compliance gaps highlighted in recent 483 observations like Torrent Pharmaceuticals. Our AI-powered platform transforms manual, error-prone validation processes into systematic, compliant, and efficient workflows that exceed regulatory expectations.
Core AI FDA Cleaning Validation Capabilities:
1. Automated Protocol Generation and Risk Assessment
- Smart protocol creation based on equipment type, product characteristics, and contamination risks
- Automatic worst-case scenario identification using API solubility, potency, and toxicity data
- Risk-based sampling location determination with statistical justification
- Cross-product contamination impact assessment using HBEL calculations
2. AI-Powered Residue Limit Calculations
- Machine learning algorithms calculate scientifically justified acceptance criteria
- Health-based exposure limit (HBEL) integration for patient safety assurance
- Statistical significance testing for cleaning effectiveness validation
- Real-time limit adjustments based on regulatory guidance updates
3. Intelligent Validation Execution and Monitoring
- Automated test scheduling and sample tracking with chain of custody
- Real-time analytical result integration from LIMS and testing systems
- Exception-based monitoring flags deviations and out-of-specification results
- Continuous validation status tracking with automated escalation protocols
4. Regulatory-Ready Documentation and Reporting
- ALCOA+ compliant data capture with complete audit trail generation
- 21 CFR Part 11 electronic signature integration for approval workflows
- Inspection-ready validation reports demonstrating scientific rigor
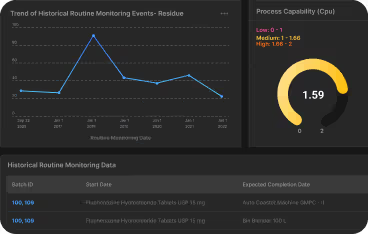
- Real-time dashboard metrics for management oversight and regulatory preparedness
How CLEEN Directly Addresses Torrent-Type FDA 483 Risks
Preventing Inadequate Cleaning Procedures:
- Automated cleaning procedure generation based on equipment design and product characteristics
- Scientific justification documentation for each cleaning step and parameter
- Validation protocol templates ensuring complete residue removal assessment
- Continuous procedure effectiveness monitoring with statistical trend analysis
Eliminating Cross-Contamination Risks:
- Comprehensive product-to-product carryover risk assessment using toxicological data
- Automated worst-case cleaning scenario identification and validation
- Equipment surface area mapping with targeted sampling location optimization
- Real-time cleaning effectiveness verification before subsequent product manufacturing
Ensuring Documentation Completeness:
- Electronic validation protocols with mandatory field completion and approval workflows
- Automated report generation meeting inspection requirements
- Version control and change management for all validation documents
- Integrated CAPA management linking cleaning failures to systematic corrective actions
Schedule a Demo to see CLEEN prevent your next FDA 483 observation.
Advanced Analytics and Regulatory Intelligence
Validation Performance Benchmarking:
- Industry comparison metrics showing validation performance versus pharmaceutical peers
- Inspection preparedness scoring based on FDA cleaning validation completeness
- Time-to-validation analytics identifying bottlenecks and optimization opportunities
- Validator performance tracking ensuring consistent quality across all protocols
Predictive Validation Analytics:
- Cleaning failure prediction modeling using equipment history and product complexity
- Validation resource planning optimizing team allocation based on protocol requirements
- Regulatory trend analysis anticipating focus areas for proactive compliance
- Cross-contamination risk forecasting preventing product quality incidents
Transform Your FDA Cleaning Validation: From Reactive to Proactive
The Business Case for AI FDA Cleaning Validation Automation
ROI Calculation for CLEEN Implementation:
Cost Savings (Annual):
- Validation time reduction: 70% faster protocol completion (60 hours → 18 hours per validation)
- Regulatory risk mitigation: Avoid $3-8M in batch holds and cross-contamination incidents
- Resource optimization: Redirect 40% of validation resources to strategic quality initiatives
- Compliance cost reduction: 60% decrease in cleaning validation-related audit findings
Revenue Protection:
- Faster product launches: Reduce validation-related delays by 75%
- Market access acceleration: Prevent 483 observations that delay approvals
- Customer confidence: Improve supply reliability through predictive validation analytics
- Competitive advantage: Achieve validation excellence differentiating your quality brand
Implementation Success Stories
Global Pharmaceutical Company Case Study:
"After implementing Leucine CLEEN, we reduced our FDA cleaning validation cycle time from 8 weeks to 2 weeks. More importantly, our recent inspection resulted in zero cleaning validation observations – transforming our biggest compliance risk into a competitive advantage."
– Director of Quality Assurance, Top 15 Global Pharmaceutical Company
Contract Manufacturing Organization Results:
"CLEEN's automated risk assessment identified cross-contamination scenarios we missed with manual validation. This prevented a potential product recall that could have cost us $20M and damaged relationships with three major clients."
– VP Quality Operations, Leading CDMO
Start Your AI FDA Cleaning Validation Transformation with a personalized demo and ROI assessment.
Getting Started: Your Path to FDA Cleaning Validation Excellence
Phase 1: Assessment and Planning (Weeks 1-2)
- Current FDA cleaning validation process evaluation and regulatory gap analysis
- Inspection preparedness assessment using industry intelligence
- Team competency analysis and change management planning
- Integration requirements with existing quality and manufacturing systems
Phase 2: CLEEN Configuration (Weeks 3-6)
- Validation workflow design optimized for your products and equipment
- AI algorithm training using your historical validation and analytical data
- LIMS and analytical system integration with data validation
- User acceptance testing and feedback incorporation across validation teams
Phase 3: Deployment and Optimization (Weeks 7-10)
- Phased rollout across equipment types and manufacturing sites
- Performance monitoring and continuous improvement implementation
- Validator training and competency verification programs
- Regulatory compliance validation and inspection preparation
Ready to Transform Your FDA Cleaning Validation Strategy?
Don't let inadequate cleaning validation become your next FDA 483 observation. Join leading pharmaceutical companies who have transformed their cleaning validation from a compliance burden into a competitive advantage with Leucine CLEEN.
What you'll get in your personalized CLEEN demo:
- Live demonstration of AI-powered protocol generation and risk assessment
- Custom ROI calculation based on your current validation processes
- Integration roadmap with your existing quality and manufacturing systems
- Regulatory compliance gap analysis and improvement recommendations
- Implementation timeline and change management strategy
Book Your CLEEN Demo Today
Transform your FDA cleaning validation strategy with Leucine's AI-powered quality management platform. Join leading pharmaceutical companies who trust Leucine to exceed regulatory expectations and drive operational excellence.
Ready to prevent your next FDA 483 cleaning validation observation? Contact Leucine today for a personalized demonstration of how CLEEN transforms pharmaceutical FDA cleaning validation from reactive compliance burden to proactive competitive advantage.
Frequently Asked Questions
How does CLEEN ensure regulatory compliance compared to manual FDA cleaning validation processes?
CLEEN is built specifically to meet 21 CFR Part 211.67 requirements and modern expectations for risk-based FDA cleaning validation. Our platform automatically ensures regulatory compliance by providing systematic protocol generation with scientific justification, AI-powered residue limit calculations using HBEL principles, automated documentation creating inspection-ready validation reports, and comprehensive cross-contamination risk assessment extending validation to all affected products and equipment. Unlike manual processes, CLEEN never misses regulatory requirements and continuously updates based on evolving guidance and industry best practices.
What ROI can pharmaceutical companies expect from implementing automated FDA cleaning validation?
Companies typically see 4-6x ROI within 12 months through multiple value streams: 70% reduction in validation cycle time (from 60+ hours to 18 hours per protocol), 85% decrease in cleaning validation-related batch delays and cross-contamination incidents, 60% reduction in 483 observations related to FDA cleaning validation inadequacies, and 40% resource optimization allowing validation teams to focus on strategic quality initiatives. For a typical pharmaceutical company with 50 cleaning validations annually, this translates to $1.5-3M in direct cost savings plus significant risk mitigation value from prevented enforcement actions and product recalls.
How does Leucine CLEEN integrate with existing pharmaceutical quality and manufacturing systems?
CLEEN is designed for seamless integration with your current pharmaceutical technology ecosystem through pre-built connectors for major ERP, LIMS, and manufacturing execution systems, API-based integration with Electronic Batch Records, analytical instruments, and equipment management systems, flexible data mapping accommodating existing validation workflows and data structures, and cloud-native architecture scaling with business needs without infrastructure investment. Our implementation team ensures zero disruption to ongoing validation activities while providing comprehensive training and change management support for validation teams across all sites.