Building a Robust Cleaning Validation Protocol: A Blueprint for 2025

As pharmaceutical manufacturing grows more complex and regulatory agencies tighten their expectations, building a robust Cleaning Validation Protocol is critical for QA, QC, and validation teams in 2025. Modern protocols are no longer just box-ticking exercises—they’re strategic tools for ensuring product quality, patient safety, and global compliance across multi-product and high-volume facilities.
What Is a Cleaning Validation Protocol?
A cleaning validation protocol is a systematic, documented strategy outlining how equipment is cleaned, sampled, analyzed, and monitored to control chemical, microbial, or cross-product contamination. In 2025, such protocols must be risk-based, lifecycle-managed, and scientifically justified—encompassing robust documentation, digital traceability, and rapid adaptation to regulatory changes.
Why Cleaning Validation Protocols Matter Now More Than Ever
Key Regulatory Drivers
- Stricter Global Standards: FDA, EMA, MHRA, WHO, and ANVISA demand validated protocols with quantifiable residue limits, health-based exposure limits (HBELs), and rigorous documentation. Refer to Cleaning Validation Guidelines 2025 for details.
- Audit Readiness: Regulatory bodies expect digital traceability, standardized records, and real-time monitoring, making outdated paper-based protocols liabilities. Use this Audit Readiness Checklist 2025 to assess your protocol.
- Operational Efficiency: Automated protocol management reduces manual errors, supports seamless audits, and enables cross-functional collaboration in QA, QC, and manufacturing.
Pro Tip: Automate residue calculations and audit documentation using Leucine’s Cleaning Validation Software (CLEEN) for FDA, EMA, and WHO compliance, minimizing non-compliance risks.
Blueprint: Building Your Cleaning Validation Protocol for 2025
- Define Objective, Scope, and Regulatory Basis
Reference all relevant cGMP, FDA (21 CFR Part 211.67), EMA, and WHO guidelines. Specify equipment, products, and the rationale for inclusion. - Risk-Based Product and Equipment Grouping
Identify worst-case scenarios—lowest solubility, highest toxicity, challenging equipment. Use HBEL and Maximum Allowable Carryover (MACO) calculations tailored to each API. - Sampling and Analytical Methods
Use validated methods: Swab sampling, rinse sampling, and modern analytics (TOC, HPLC, LC/MS). Define and document sampling locations; regulatory focus includes both product-contact and non-contact surfaces. - Setting Acceptance Criteria
Calculate residue limits using HBEL, PDE, and MAC. Agencies no longer permit visual inspection alone—statistically justified, quantifiable limits are required. Refer to Cleaning Validation Guidelines 2025 for calculation methodologies. - Cleaning Procedures & Parameters
Standardize cleaning agents, cycles, critical parameters (time, temperature). Include change management, deviation handling, and campaign strategies. - Validation Lifecycle and Digital Monitoring
Treat validation as continuous: periodic and trigger-based revalidation is a must. Track trend data digitally; use CAPA systems for deviations. - Documentation, Reporting, and Audit-Readiness
Use version-controlled digital templates (SOPs, reports, deviation logs). Integrate with LIMS and automate audit trails to ensure inspection consistency.
2025 Best Practices & Emerging Trends
- Digital Transformation and Automation
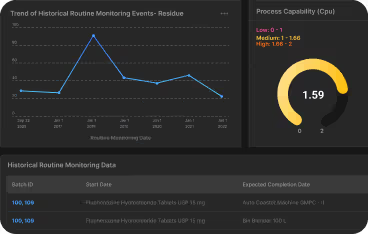
Automated cleaning validation platforms eliminate manual calculations and ensure data integrity. Digital tools can reduce validation cycle times and speed up deviation detection. - Risk-Based & Lifecycle Approaches
Focus resources where risk is highest; adapt protocols to new products, process changes, or deviations. Statistical trending and process capability monitoring are expected by regulators. - Health-Based & Data-Driven Limits
Use toxicological data and historical trends for residue limits, especially in multi-product facilities. HBELs and PDEs are now industry standards, replacing arbitrary limits. - Cross-Functional Collaboration
Build protocols and corrective actions with input from validation, QA, QC, manufacturing, regulatory, and training. - Harmonized Global Standards
Align your master plan to FDA, EMA, MHRA, WHO, and ANVISA requirements for smoother audits and multinational compliance.
Deep dive into the Ultimate Guide to Cleaning Validation – White Paper 2025 for comprehensive insights.
Pro Tip: Save time with CLEEN: automate protocol generation, risk scoring, and validation scheduling across global facilities.
Leveraging CLEEN Cleaning Validation Software
CLEEN automates:
- HBEL-based residue limit calculations
- Audit-ready validation documentation
- Instant risk assessment for new drug introduction
- Integration of residue and status data from LIMS
- Real-time dashboards for validation status and deviations


Pro Tip: CLEEN’s real-time monitoring and instant audit reporting keep you one step ahead of regulatory expectations.
Frequently Asked Questions
1. What is the difference between a cleaning validation protocol and a SOP?
The protocol defines the overall approach and acceptance criteria for cleaning validation, while an SOP details step-by-step procedures for cleaning execution.
2. Why are HBELs and MACO calculations essential for 2025?
They scientifically justify residue limits using toxicological and pharmacological data, enabling safer cross-contamination risk management compared to old visually-based standards.
3. How does automation improve cleaning validation?
Digital solutions reduce errors, ensure up-to-date documentation, and facilitate rapid audits, enabling focus on high-risk areas and continuous process improvement.