



Top 5 Subsystems in 2025: Highest Impact Areas in FDA Inspection Preparation
Struggling with FDA inspection preparation? Discover the top 5 subsystems that trigger the most citations, and how to avoid them with FDA Tracker insights.

As FDA inspections evolve and grow more data-driven in 2025, manufacturers must sharpen their inspection readiness by focusing on the systems that attract the most scrutiny. In the context of regulatory inspections, it is essential to be prepared for both scheduled and unscheduled visits, as inspections may occur with advance notice or without any notice at all. Based on the latest data from FDA Tracker, five subsystems stand out for their high number of Form 483s and Warning Letters: Contamination Control, Environmental Monitoring, Cleaning Validation, Document Management, and Investigations.


A proactive approach to FDA inspection preparation involves understanding the inspection process, relevant regulations, and the importance of regulatory compliance. It is also critical to understand FDA regulations and ensure your organization meets all of FDA's requirements as part of being fully prepared for regulatory inspections. Regulatory affairs teams and a dedicated inspection readiness team play a critical role in coordinating preparation efforts and ensuring compliance across departments. Contract research organizations and clinical research teams are also key stakeholders in inspection readiness, especially in regulated environments.
If you’re in charge of FDA inspection preparation, these are the key areas you cannot afford to overlook for inspection readiness.
During an FDA inspection, organizations can expect a thorough review of documentation, processes, and compliance with FDA's requirements. Being prepared in advance is essential for a successful inspection, whether or not you receive advance notice or any notice at all. Continuous readiness ensures your organization can respond effectively to both announced and unannounced regulatory inspections.

1. Contamination Control
Data till 2025: 192 Form 483s | 25 Warning Letters | Total: 217
Contamination Control leads the list once again. This subsystem directly affects patient safety, particularly in sterile and aseptic environments. The FDA is closely monitoring cross-contamination risks, environmental control gaps, and poorly maintained cleanroom facilities. Maintaining a well-organized and operationally ready facility, supported by a robust quality system and comprehensive quality systems, is essential to support effective contamination control and demonstrate a commitment to product quality.
Common Pitfalls:
- Inadequate environmental zoning
- Poor airflow validation
- Insufficient gowning practices
- Failing to address potential issues in critical processes and procedures that impact the entire facility and product quality
- Lack of clearly defined responsibilities among team members during contamination control audits, which can hinder effective preparation and response
- Inspectors will closely review contamination control processes and the operation of cleanroom facilities to ensure compliance
Action Step: Mock audits should be conducted regularly, focused on contamination risks. Regular practice through these mock audits ensures your team is prepared for real inspections. FDA Tracker helps you review historical data and trending observations so your team knows exactly what to fix before the FDA arrives.
Explore FDA’s Six-System model with real-time risk ratings and insights. Explore now
2. Environmental Monitoring
Data till 2025: 170 Form 483s | 20 Warning Letters | Total: 190
The Environmental Monitoring subsystem has seen a sharp uptick in inspection observations, especially for facilities producing sterile injectables and biologics. Routine trending, alert limit excursions, and lack of follow-up CAPAs (corrective and preventive actions) are among the top cited issues.
FDA inspection preparation for this subsystem should prioritize:
- Active air sampling frequency
- Microbial trending and response protocols
- Training of staff in viable sampling techniques
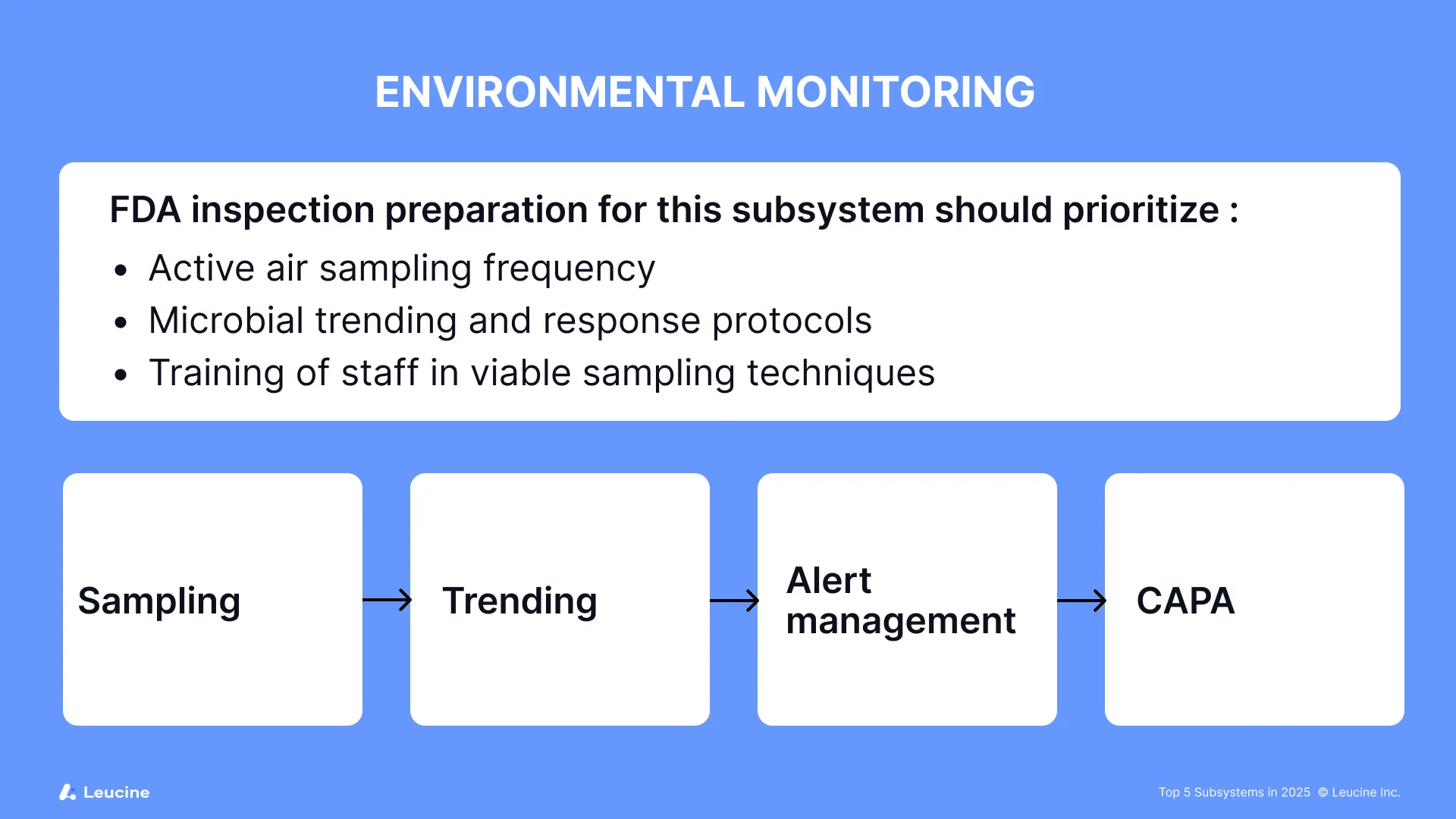
Issues identified during monitoring should be addressed promptly to maintain compliance. Conducting thorough risk assessments and adopting a risk based approach to environmental monitoring are critical for identifying vulnerabilities and guiding corrective actions. Regular internal audits play a key role in identifying compliance gaps in environmental monitoring and supporting continuous improvement. Robust processes and well-managed operations, supported by strong quality systems, are essential for ongoing compliance, ensuring that environmental monitoring activities remain effective and inspection-ready. Organizations should be prepared for inspection findings related to environmental monitoring by having comprehensive CAPA plans in place.
Staff training should include regular practice through mock inspections and exercises to ensure staff are inspection-ready and confident in their roles.
Pro tip: FDA Tracker breaks down trends in Environmental Monitoring citations so you can prioritize preventive actions before your next inspection.
3. Cleaning Validation
Data till 2025: 131 Form 483s | 19 Warning Letters | Total: 150
Cleaning Validation continues to be a complex and heavily cited subsystem. Whether it’s missing residue acceptance criteria or inadequate sampling points, FDA expects statistically justified cleaning protocols that ensure patient safety and product integrity. Training should be grounded in good clinical practice principles to ensure staff understand regulatory requirements and compliance expectations.
Watch for:
- Lack of dirty-hold time validation
- Insufficient visual inspection training
- Non-detectable swab methods
- Incomplete or outdated procedures and missing training records
Regular practice through training exercises is essential to ensure your team is prepared for inspections. Any issues identified or finding of gaps during cleaning validation reviews should be documented and addressed. Corrective and preventive actions (CAPAs) must be implemented as part of the cleaning validation process to resolve non-compliance and prevent recurrence.
4. Document Management
Data till 2025: 120 Form 483s | 20 Warning Letters | Total: 140
Missing, outdated, or unsigned documents continue to haunt manufacturers. With data integrity being a core focus area, any gaps in SOP version control, logbook completion, or audit trail management are likely to attract citations. During inspections, the ability to quickly find and provide requested documents is critical. The front room serves as the controlled space for inspector interactions, where the FDA inspector, FDA investigator, or FDA investigators will review documentation and may ask you to answer questions about document control. The back room or war room acts as the command center for tracking every document request, organizing responses, and ensuring smooth document flow. FDA field investigators also play a key role in reviewing document management practices to ensure compliance with regulatory requirements.
Checklist for FDA inspection preparation:
- Ensure real-time review and approval of key GMP documents
- Maintain centralized access to batch records and quality manuals
- Validate electronic systems used to manage documentation
- Ensure quick access to requested documents and track every document request during inspections
- Assign clear responsibilities for document retrieval and response during inspections
- Be prepared to provide documentation and respond to inspector inquiries
- Conduct regular practice drills for document management to ensure readiness
Following guidance on document management best practices, supported by a robust quality system, can help prevent common pitfalls and demonstrate compliance. Any issues or gaps identified during document review should be addressed promptly to avoid negative findings during inspections.
📂 Explore the Document Management Dashboard on FDA Tracker to analyze warning letter language, common triggers, and prevent repeat issues.

5. Investigations
Data till 2025: 112 Form 483s | 21 Warning Letters | Total: 133
Poor root cause analysis and inadequate corrective and preventive actions (CAPAs) were cited in nearly every major Form 483 issued under this subsystem. FDA wants to see that manufacturers can identify systemic issues and implement sustainable fixes. Addressing root causes and implementing effective corrective and preventive actions in response to inspectional observations and findings is critical for demonstrating compliance and inspection readiness. Issues identified during investigations should be thoroughly documented and addressed as part of a robust quality system.
Inspection preparation tips:
- Use fishbone diagrams or 5-Whys for every deviation
- Validate CAPA effectiveness with clear closure evidence
- Train teams on distinguishing between superficial and systemic failures
- Prepare thorough responses to inspection findings, identify gaps in investigations, and ensure all inspectional observations are systematically addressed
- Regularly practice investigation techniques to improve team performance and ensure staff are prepared for inspection questions related to investigations
- Clearly define responsibilities among team members during investigations to ensure efficient and effective processes
- Ensure your quality systems support comprehensive investigations, documentation, and continuous improvement
FDA Tracker’s Investigations module offers real-world examples of failed investigations—and what effective ones look like.
Are You Focusing on the Right Subsystems?
These five areas account for the majority of inspection findings in 2025. Yet many teams spread their audit prep thin across low-risk zones.
FDA Tracker helps teams prioritize where it matters most—with real-time data on inspection frequency, severity, and enforcement trends across subsystems.
Ready to strengthen your inspection prep strategy? Sign up for free on FDA Tracker and turn inspection data into daily readiness.
